Hi John,
Looking for some guidance...
I’ve written a matrix correction program (in Fortran 90/95) so that I can re-process output from the JEOL PC-EPMA software, as I don’t necessarily like the options offered by JEOL. Where possible, I’ve been testing output from my program against output from CalcZAF.
For the PAP, XPP, and FRAME-ZAF models, I can duplicate f(chi) calculated by CalcZAF precisely for X-rays in both pure elements and mixtures. However, I’m having problems doing the same using the Armstrong absorption model. I’ve attempted to locate the source(s) of the discrepancy by combing through the code that you’ve posted on-line, primarily in the file ZAF.bas. However, I have no substantive knowledge of Visual Basic syntax or program structure, and so I’ve had some difficulty following equations through the code; for instance, I can’t seem to find exactly where the integrals of phi(rho*z) and phi(rho*z)*(-chi)*rho*z are evaluated. Could you point me in the right direction?
When I calculate f(chi) for K lines of pure elements using the Armstrong absorption model, I get exactly the same output as generated by CalcZAF. However, for K lines of elements in mixtures, my f(chi) results differ from the CalcZAF output (example below). In addition, when I calculate f(chi) for M lines of pure elements, my output always differs at least slightly from the CalcZAF output. For instance, for W Ma in pure W, I get f(chi) = 0.6798, while CalcZAF gives 0.6727 (accelerating potential = 15 kV, takeoff angle = 40 degrees, MAC database = MAC30). For Ce Ma in pure Ce, I get f(chi) = 0.5158, while CalcZAF gives 0.4969. For L lines of pure elements, I get f(chi) that differs by no more than 0.0001 from CalcZAF output.
For the case of the K lines in mixtures, the discrepancy could not be due, for instance, to use of a different “mixing rule” for the mean ionization energy. This would impact the calculation of stopping power as well, but the value that I calculate for this latter quantity in the mixture is identical to the value calculated by CalcZAF.
The expressions below (which I’ve proofread many times) are consistent with those presented in Armstrong’s paper in Electron Probe Quantitation, though his presentation isn’t really complete (for instance, he doesn’t specify the Berger-Seltzer expression for mean ionization energy). I’ve replaced the ambiguous E in the expression for alpha with E0, and this appears to be consistent with the equation given in line 1130 in ZAF.bas.
Regarding my calculation of f(chi), I’ve checked results from the analytical expressions that I give below against results obtained using Simpson’s rule (via subroutine qsimp from Numerical Recipes).

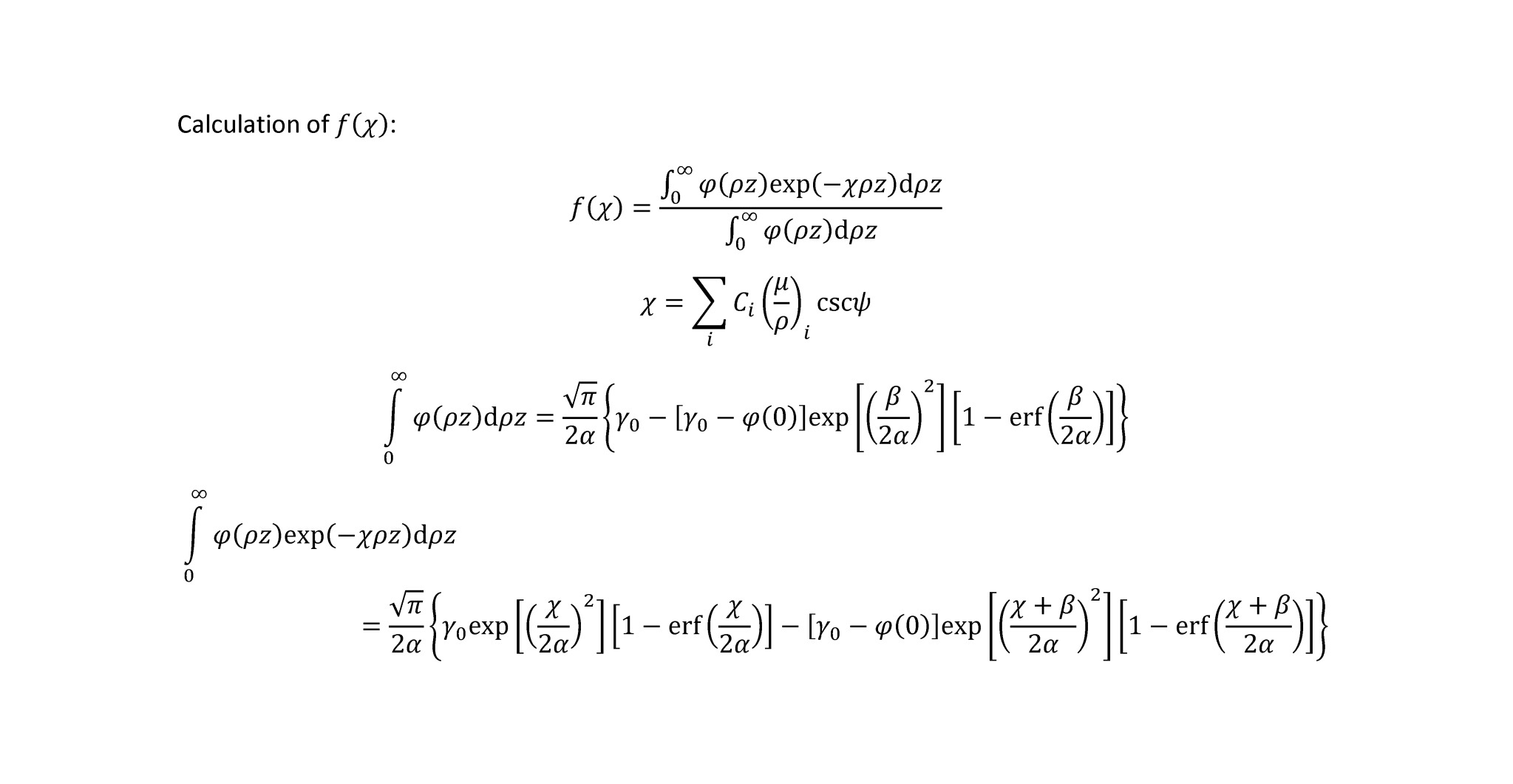
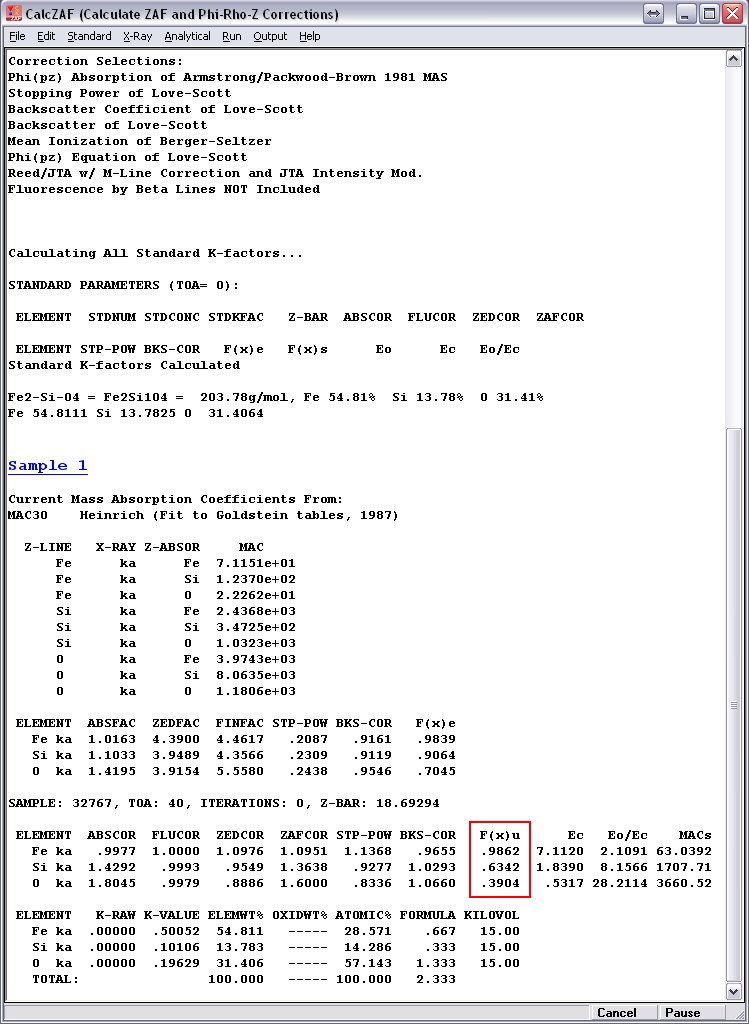
Here I give an example of the differences in my calculated f(chi) versus those from CalcZAF for the case of Fe2SiO4 analyzed at 15 kV potential and 40-degree takeoff angle. Some very minor discrepancies are present in the MACs (which I’ve calculated using Heinrich’s MAC30 equations), but these differences are too small to have any impact on calculated f(chi) at the illustrated four digits of precision. For Si Ka in the mixture, I calculate f(chi) = 0.6303, while CalcZAF gives 0.6342. For O Ka in the mixture, I calculate f(chi) = 0.3861, while CalcZAF gives 0.3904.
CalcZAF output:
My output:
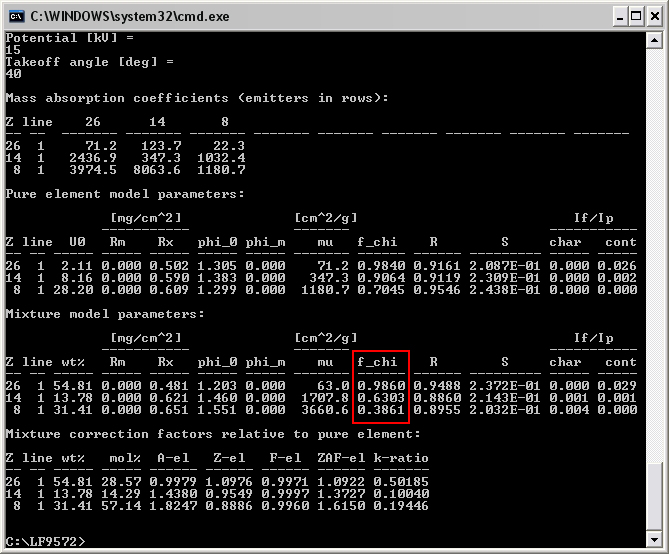
Any thoughts? Would you be willing to let me have a look at the older CITZAF code, which might be easier for me to pick through? I’ve been tearing my hair out trying to figure out why I’m getting different results.


 To post in-line images, login and click on the Gallery link at the top
To post in-line images, login and click on the Gallery link at the top