Recently I attempted to acquire more k-ratios in order to extract effective take of angles for my various spectrometer/crystal combinations. Initially I utilized Si Ka at 25 keV on SiO2 as a primary standard and various silicate such as Fe2SiO4 and Ni2SiO4 as secondary standards. The results can be seen in this topic here:
https://probesoftware.com/smf/index.php?topic=1569.msg12177#msg12177One observation from these measurements was that the measured k-ratios for TAP crystals appeared to be higher than one would expect from modeling (and when compared to the EDS detector which one might assume would have a more accurate takeoff angle since it contains no moving parts), possibly indicating a lower effective take off angle than the nominal value of 40 degrees. While the PET crystals appeared to produce k-ratios lower than one might expect, perhaps indicating a higher effective take off angle than expected.
But in the case of the TAP crystals the Si Ka peak position is very close to the lower limit, while on a PET crystal the Si ka peak position is very close to the lower limit, so to test whether this divergence in the effective take off angle from the nominal take off angle (40 degrees) is consistent over the entire range of the spectrometer, I decided to try another emission line starting with something that would be closer to the other end of the TAP spectrometer range, for example F Ka.
So I decided to try measuring F Ka k-ratios on my two fluoride standards, CaF2 (natural) and BaF2 (synthetic). Since the concentration of CaF2 is much higher I decided to make that the primary standard (as one does) and BaF2 the secondary standard:
f = 21.670 835 BaF2 (barium fluoride)
f = 48.800 831 Fluorite U.C. #20011
What I did not realize until later was that due to an extremely large absorption of F Ka by Ca, the intensity emitted from BaF2 is actually higher than CaF2!

So the k-ratios are greater than one, but for the purposes of determining effective take off angles of the TAP crystals at a high sin theta, this should not be an issue. I will report on these k-ratio measurements in the effective take off angle topic linked at the start of this post later on, but in the meantime I want to discuss something else related to APFs which is the subject of this topic...
What I also did not expect was that there appears to be a very large peak shape change between CaF2 and BaF2. I'm guessing that this peak shape change is due to the very large absorption in CaF2 rather than any sort of valence bonding differences given that both compounds are alkali earth fluorides, but please chime in if any of you has looked into this at all.
Here are the various MACs for F Ka in Ba:
MAC value for F Ka in Ba = 3155.43 (LINEMU Henke (LBL, 1985) < 10KeV / CITZMU > 10KeV)
MAC value for F Ka in Ba = 3140.00 (CITZMU Heinrich (1966) and Henke and Ebisu (1974))
MAC value for F Ka in Ba = .00 (MCMASTER McMaster (LLL, 1969) (modified by Rivers))
MAC value for F Ka in Ba = 3232.77 (MAC30 Heinrich (Fit to Goldstein tables, 1987))
MAC value for F Ka in Ba = 7408.73 (MACJTA Armstrong (FRAME equations, 1992))
MAC value for F Ka in Ba = 2913.02 (FFAST Chantler (NIST v 2.1, 2005))
MAC value for F Ka in Ba = 3160.00 (USERMAC User Defined MAC Table)
Although there is one outlier, the MACs for F Ka in Ba are all in fairly close agreement and also not terribly large values. However, the MACs for F Ka in Ca are much higher and surprisingly are pretty much in agreement (with Armstrong again a bit a an outlier):
MAC value for F Ka in Ca = 12415.20 (LINEMU Henke (LBL, 1985) < 10KeV / CITZMU > 10KeV)
MAC value for F Ka in Ca = 12370.00 (CITZMU Heinrich (1966) and Henke and Ebisu (1974))
MAC value for F Ka in Ca = .00 (MCMASTER McMaster (LLL, 1969) (modified by Rivers))
MAC value for F Ka in Ca = 12623.93 (MAC30 Heinrich (Fit to Goldstein tables, 1987))
MAC value for F Ka in Ca = 14236.12 (MACJTA Armstrong (FRAME equations, 1992))
MAC value for F Ka in Ca = 12132.31 (FFAST Chantler (NIST v 2.1, 2005))
MAC value for F Ka in Ca = 12400.00 (USERMAC User Defined MAC Table)
Looking at wavescans on both materials we see this:
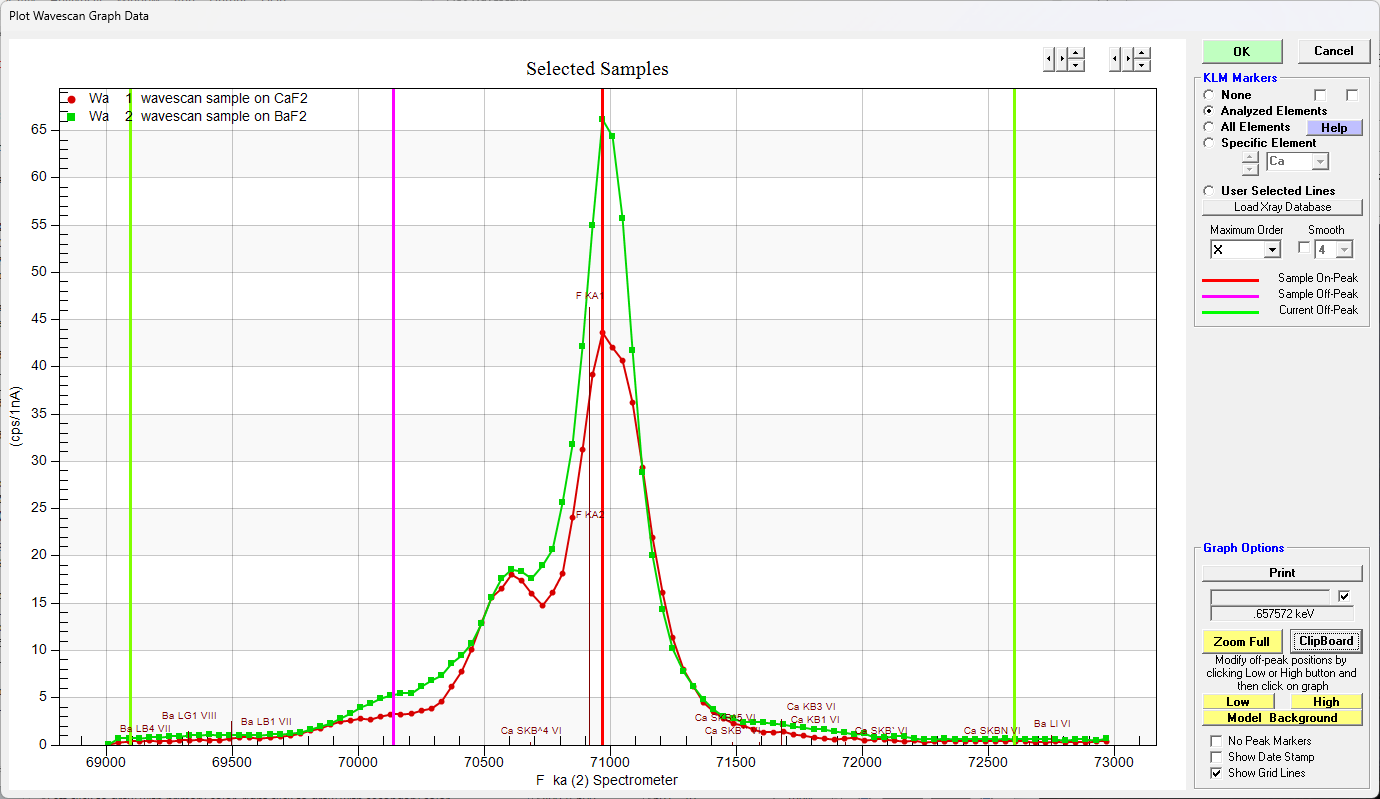
Clearly the peak intensities do not represent the integrated intensities. And in fact we can see this quite clearly when we examine the k-ratios from the EDS spectrometer and compare them to the WDS spectrometers as seen here:
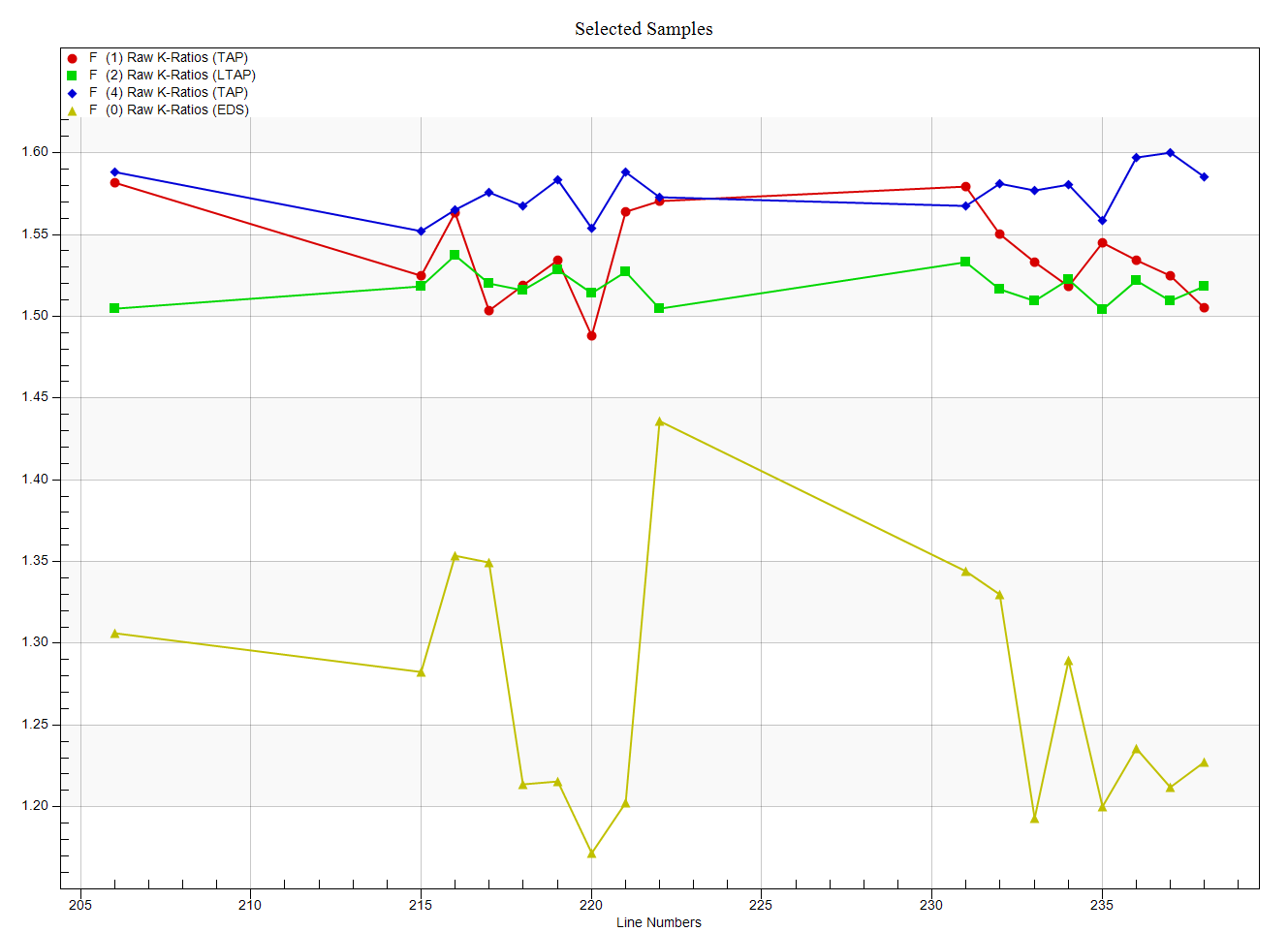
Admittedly not a great dataset, but the fact that the EDS and WDS disagree so much, I thought it might be worth looking at the Area Peak Factors (APFs) for this system a bit more. Also note that the peak position for F Ka for these two materials has not shifted, but only the peak shape. So lets start with the CaF2 (our primary standard) using the Model Background dialog and clicking the Integrate button as shown here:
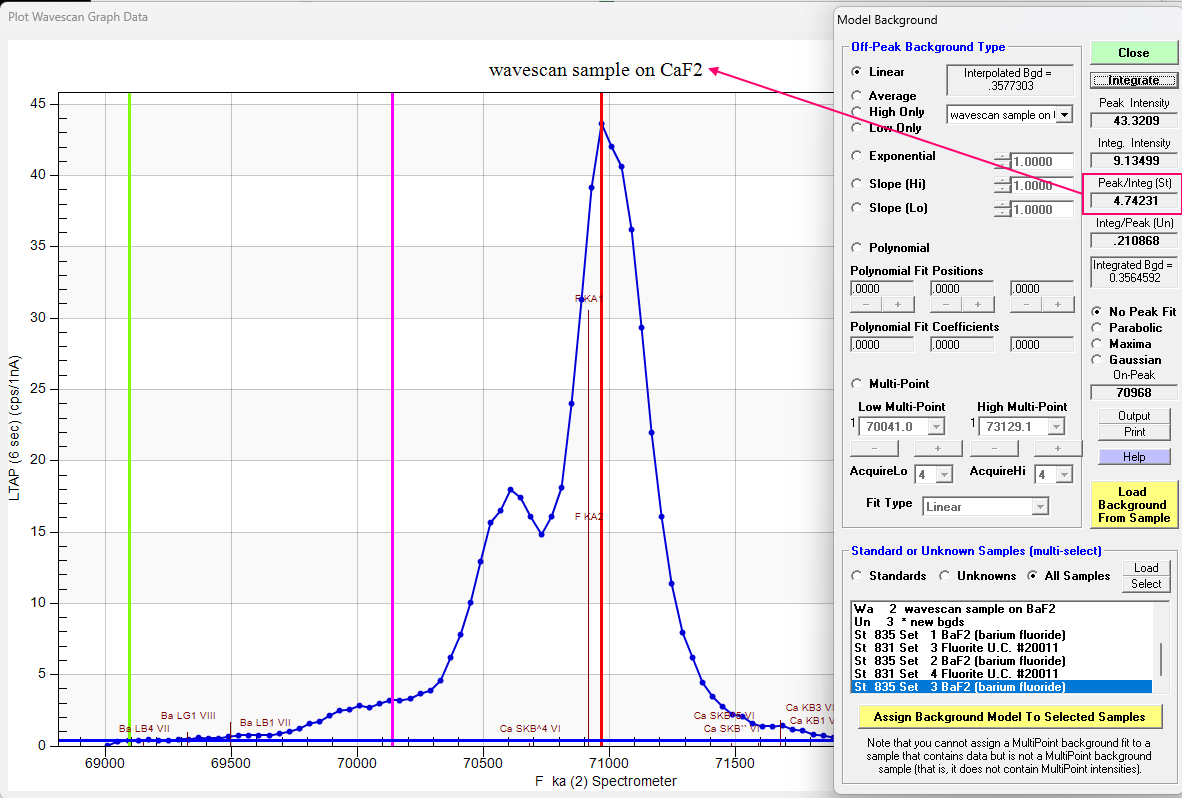
Note that we utilize the Peak/Integrated ratio for the primary standard (CaF2) and for the BaF2 (our secondary standard) we have:
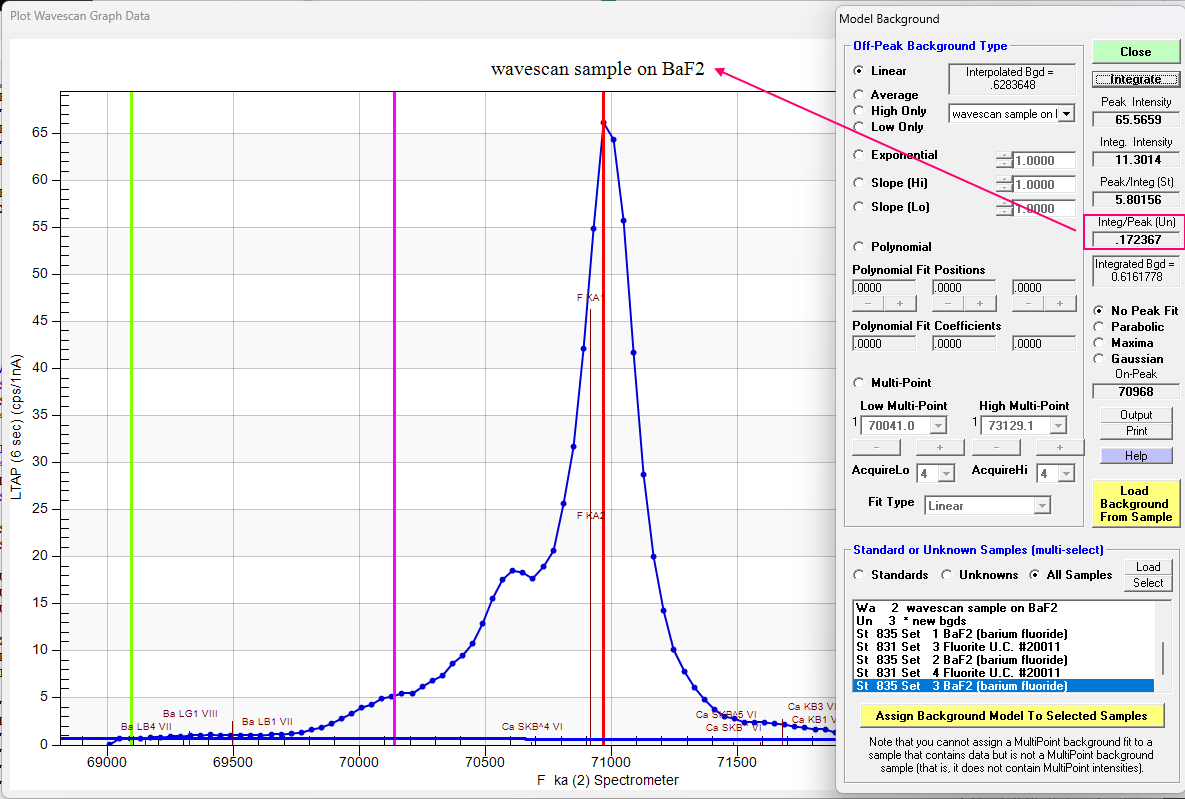
Multiplying these two values together we obtain ~0.817 or almost a 20% correction! Applying the values calculated for each spectrometer (all three are close to ~0.80) by entering these "specified" APFs in the Elements/Cations dialog as discussed in this topic and elsewhere, we obtain the following k-ratios (the APFs are applied by Probe for EPMA automatically to the k-ratios during the matrix correction):
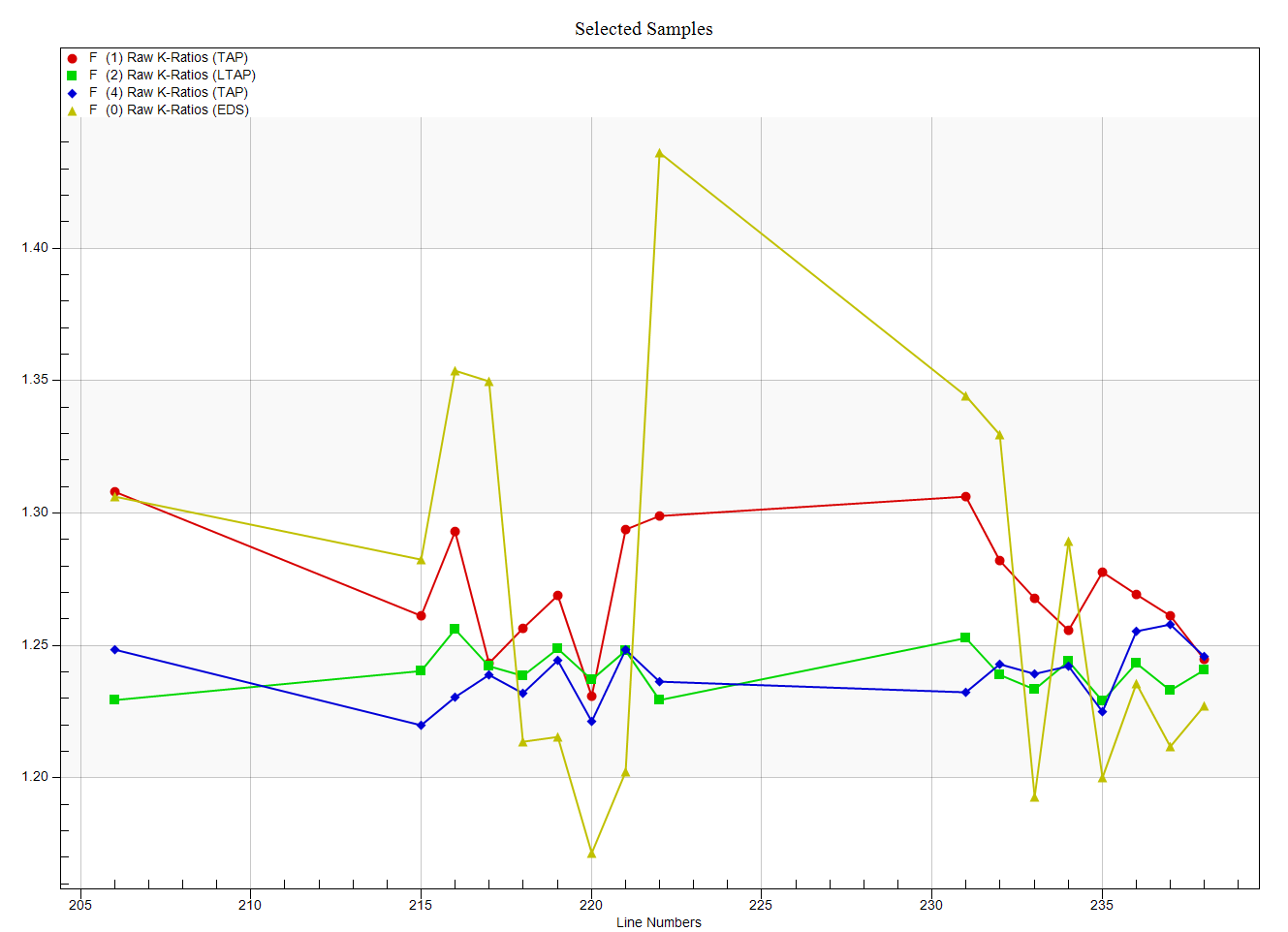
Which is a considerable improvement over what we had from assuming that the F Ka peak shape was unaffected by the emission matrix. Sorry for the digression, but I thought it was an interesting system to test the APF correction on...
So what does this mean for the analysis of F Ka? Well, given this enormous peak shape correction but also the success of modeling the peak shape using APFs, one could say to use either material as the standard and use the APF correction relative to the unknown in question, as a specified APF.
But maybe it is more accurate is to use CaF2 as a primary standard for characterizing Fluor-apatites (since there is a large amount of Ca in the apatite matrix), and use BaF2 as a primary standard for fluorine in other (less absorbing) matrices.
What do you all think?
Oh, and by the way, there is a quite large TDI correction for these materials even using a 20 um defocused beam! I will show that data in another topic also later on because I am seeing something quite odd there (what appears to be a significant difference in the TDI correction for each of the TAP spectrometers!).


 All Electron Probe Micro-Analysts are welcome to register and post!
All Electron Probe Micro-Analysts are welcome to register and post!