I just want to point that not only non-existent pile-up correction (which probeman keeps calling the "dead-time" calibration), but also some nasty 2nd, 3rd or 4th order emission lines of overlooked and not corrected overlaps (especially on the background) had produced some classical matrix matched standard cargo cultism. Don't understand me wrong, this new pile-up correction log equation (or as probeman calls "dead-time calibration") is one of the most important recent advancements (I am looking forward to see the log equation for which I have my own physical model and explanation why it works and why it stops working at very high counting rates, and it does not require physical contradictions). However, even without that, if using manufacturers recommended dead-time values (the real blanking timing, that is 3.3µs for all of Cameca WDS) and staying at count rate where pile-ups do not exceed 0.5% of registered pulses (basically below 10kcps on Cameca instruments) I came to conclusion that matrix matched standards can be ditched in most of the cases. Having no builtin logarithmic pile-up correction equations in Peaksight in some very rare cases I practice something what could be called "counting-rate matched simple standard measurements".
Hi SG,
I appreciate your support and comments. Thank-you.
Yes, I agree completely that much of the reason people feel the need to resort to matrix matched standards is due to a multitude of factors, among them poorly calibrated dead time constants, uncorrected/unrecognized spectral interferences, and spectrometer/crystal misalignments. I probably should write up a summary of all of these instrument calibration issues someday, for example picoammeter non-linearity could contribute to errors measuring ones standards and unknowns at different beam currents... the list goes on.
I will share the log dead time expression soon as we are currently writing it up now, but it suffices to say that the multi-term expression, described in this post, gives almost identical results to the log expression:
https://probesoftware.com/smf/index.php?topic=1466.msg11032#msg11032And as you also point out, by relying on matrix matched standards we have introduced another error and that is: do we really know exactly what are the compositions of our natural standard materials? We already know from studies by John Fournelle and Ed Vicenzi that these natural materials routinely utilized by many labs are heterogeneous and full of inclusions. This is exactly the impetus for our FIGMAS efforts with Will Nachlas and Aurelien Moy to identify high purity synthetic mineral candidates for eventual wide scale (global) distribution to microanalysis labs.
https://probesoftware.com/smf/index.php?topic=1415.0As you can tell, I am usually not fastidious when it comes to nomenclature, but whether it's called dead time or pulse pileup or photon coincidence I also agree this is an area that most labs are not paying much attention to. Frankly I never gave dead time calibrations much attention until John Fournelle, Anette von der Handt and I started performing these "constant k-ratio" measurements and we started seeing problems at moderate to high count rates on our respective instruments:
https://probesoftware.com/smf/index.php?topic=1466.msg11025#msg11025 Hopefully both Cameca and JEOL will implement these improved dead time corrections in their OEM software soon. As you point out this is especially important for Cameca instruments with their rather long dead times constants. The good news is that the current version of Probe for EPMA now contains *four* dead times corrections!

They are: the traditional (single term) expression, the Willis (1992) (two term) expression, the new six term expression, and now the logarithmic expression.

There are X-ray lines which requires matrix matched standards such as i.e. Sulphure Ka due to line shift (although if offseting the S Ka position of unknown to the correct for unknown position the sulfate-sulfide S Ka standards can be nearly interchangeable). But line shifts are actually only the half of the problem.
Yes, exactly. We always adjust our sulfur peak positions based on the average oxidation state of our volcanic glasses and just use pyrite as the primary standard as it is about a 10% effect in the case of most basaltic glasses. This peak shift effect could also be dealt with using an APF correction as well.
Sulfosalts in general are difficult beast: common extreme pace of oxidation of surface is very often overlooked and I know some laboratories uses commonly Chalcopyrite and Pyrite as main standards for sulphure (Those are often included in most of buy-able sets of mineral standards), which are a very terrible choose (I need often to deal with users coming with terrible measurement protocols designed elsewhere). Unless the unknown sample of sulfides had spent exactly same time in the same environment as standards (and we still need to suppose that unknown and CuFeS or FeS2 had oxidized at the same pace) there will be kind of correct matrix matched S-standard hunt (my bitter experience from first years of my probe job, when I was not questioning any already settled practices). Also, who re-polishes the standard-sets every week? From sulfides, AFAIK, only ZnS is resilient to oxidation in some sane degree (can withstand half year without re-polishing), and quantification works correctly if unknown is just freshly polished and ZnS is used as S Ka standard. But there comes the common problem: often user who own the samples with unknowns of sulfides is lazy and comes to lab with samples polished two weeks or few months ago... and then there is surprise Pikachu face why analytical totals are closing only to 96%. Because of laziness at least for sulfides people leaned-on "matrix matched standards" (which in correct form should be actually called "surface oxidation matched standards"). When using oxidation-resistant sulfide standard and re-polishing the unknown sulfides same or a day before probe session - the need for matrix matched standard just is no more.
So Metal vs Oxides has very similar problem of oxidation like sulfides, just a bit less severe. At least from my experience if I use metal oxide as standard, metals as unknowns always produces lower sums than expected - never the larger, which taking into consideration of oxidation of metal surface makes complete sense (also there is these O Ka peaks in WDS and EDS if looked for). The effect can be minimized by re-polishing. But perfect metal surface preparation system would require polishing, drying and transportation under the inert gas. Fortunately many metals have this feature of initial oxidated layer of the metal preventing/slowing down further oxidation, which lots of sulfides do not posses.
I agree completely about these surface oxidation (and often overlooked) effects. This is the primary reason I have insisted to Will and Aurelien that we mount these FIGMAS standards in an acrylic mount that can be easily re-polished and re-coated. For example:
https://probesoftware.com/smf/index.php?topic=172.msg8991#msg8991Now with silicates and oxides this is not so much a problem, but as you point out for metals and sulfides we find it necessary to re-polish and re-coat every few months or whenever we get sulfide work. That said, we often avoid some of these issues by running our standards and unknowns at 20 or 25 keV which reduces surface sensitivity. But for those performing low voltage work, re-polishing of standards (and unknowns) just prior to analysis is essential.
Then there is another case with complex minerals and substances which in particularly with large diff XTALs leaves no space for precise background measurement (but believe me some minerals with 30+ major and minor elements will cause a hell even with standard (small) sized diff crystals). And when if people can't get right measurements using classical approaches they turn to matrix matched standards (which have the similar troubles with background, thus the problem is partly eliminated). Ironically while from one side PfS development in its own way uncovers redundancy of matrix matched standards (i.e. by this pile-up/"dead-time" callibration) on the other hand PfS-implemented multi-point-background for sure will keep some to matrix matched standard preferences (I am fiercely opposed to the MPB and see it not only highly redundant and unnecessary, but also dangerous as results could insert some biases to large sets of inter institutional measurements). However with proper background measurement (single background with ultimately universally precise slope (single and same slope for very different density materials at same background position, often it is possible to make it without any absorption edge in between it and the peak!), which I find using HussariX software) - simple standards just work without any need of matrix matched ones. Sometimes The complex minerals are so complicating the measurements of background that it is better to switch some measurements to 2nd order lines or if applicable use the higher resolution lower intensity equivalent on other diff XTALs. (i.e. Si Ka measure on PET instead of overcrowded TAP). Fortunately recently even those cases got much easier to deal with starting with Cameca Peaksight 6.5, where negative interference corrections just works (that is background position interference with other peaks can be itteratively corrected).
F is problematic if measured on TAP, as there is F energetic dispersion depending from the type of mineral, and so TAP is too high resolution, but going to PC0 allows to use single F rich substance universally. So, what is the problem with Bastnesite if F is measured with PC0 and F is calibrated on pure F-Flogopite or pure F-Topaz? Well due to strong and wide REE M lines and if using MPB or Two (left-right) backgrounds - the situation would be just pure doom. But single background with precise slope and few precise interference corrections makes it work without any issues. As for lower atomic numbers probably some of matrix matched standards would find its niche there, I have not much experience there.
I totally appreciate the single point slope background and it is implemented in PFE as you can see here for both sides of the peak:
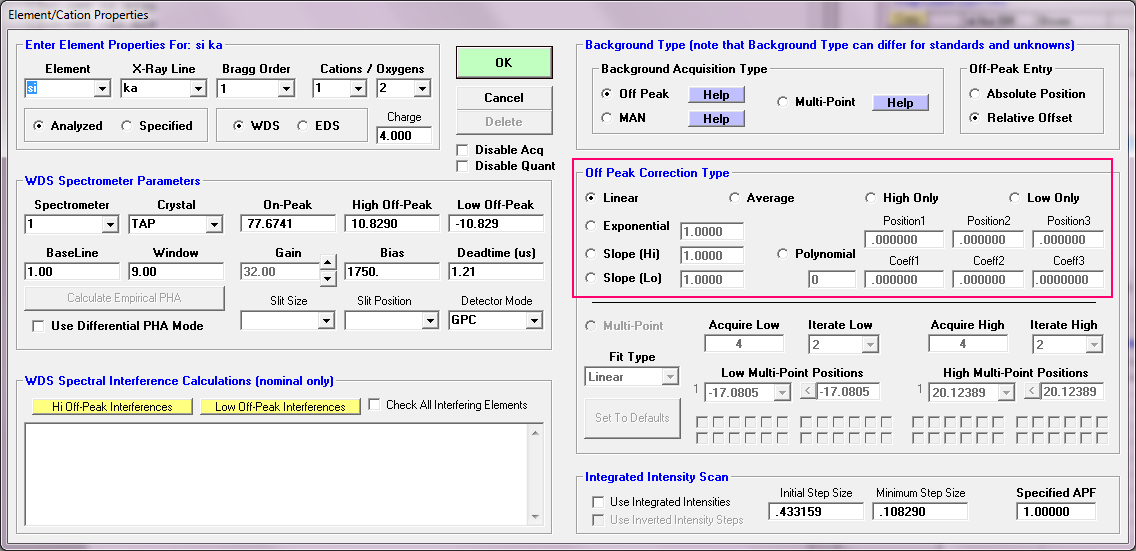
Note the Slope (Hi) and Slope (Lo) methods.
As for the MPB method, I think that it can be useful, but can also be abused like most things. However, according to Mike Jercinovic and others, it is essential for high accuracy trace elements in complex matrices. Frankly I personally prefer the MAN method where the background is calibrated at the emission line position. This method is accurate to a few hundred PPM or better in most silicates and oxides and one can further improve accuracy using a quantitative blank correction. See Donovan et al., (2011) for the blank correction and Donovan et al., (2016) for details on the MAN correction. I like particularly that one can obtain higher sensitivity in less time than off-peak methods. It's sort of like magic!

As conclusion, Every time I come across any inherited need for matrix matched standard for common rock forming or accessory minerals I can find the hidden overlooked reason behind, which often originated either due to unaccounted sample oxidation, or poor measurement practices (or both). Often it is that no one cared to look deeper as using matrix matching standard was the least-effort approach for short-term to get the predicted results. People are keen to take protocols which are known just to work, even if it is not the best or more tedious way in bigger picture. Ditching matrix matched standards is so much time saving in the long term as keeping the limited number of simple standards updated is more efficient and easier than standard base with all kind of combinations of matrices.
I could not agree more with you. Thanks for sharing your thoughts on this.


 We are a community of analysts, that cares about EPMA
We are a community of analysts, that cares about EPMA