While taking a break from synthesis of Cs compounds as I wait to see if I can come up with a platinum crucible, I’m pursuing synthesis of other compounds that could be of use as standards. When I think about my standards collection and weak spots that exist in it, it occurs to me that I don’t have much in the way of oxidized Co and Ni compounds (let alone sulfides). I prefer not to use metals as standards when analyzing oxides or sulfides because 1) oxide films can form on them very rapidly, and 2) use of them can lead to large atomic number corrections. Vanadates are attractive because 1) they can be grown at relatively low temperature and 2) vanadium is also a relatively weak spot in my standards collection. I’ve successfully grown Co
3V
2O
8 and Ni
3V
2O
8 in very simple synthesis runs, though I don’t necessarily recommend the approach that I took. Each compound is the most Co- or Ni-rich compound in the respective binary V
2O
5-bearing system; this is convenient because each can be grown by saturating the system in CoO or NiO, respectively. Co
3V
2O
8 contains 55.28 wt% CoO and 44.72 wt% V
2O
5, while Ni
3V
2O
8 contains 55.20 wt% NiO and 44.80 wt% V
2O
5.
In each case I loaded an alumina crucible with powdered V
2O
5 and an excess of either CoCl
2·6H
2O or NiCl
2·6H
2O (and this is the part that I don’t necessarily recommend). After allowing the hydrated chloride to melt and then dehydrate for a couple hours in the furnace at about 200ºC, I then ramped the temperature up to 950ºC. For the case of the Co
3V
2O
8 synthesis, I had hoped that the excess chloride would serve as a flux that could be dissolved away easily with water after the run, as anhydrous CoCl
2 melts at 735ºC. In reality the reaction to produce the vanadate proceeded violently in the vicinity of 700ºC (noting that V
2O
5 melts at 690ºC). Not only was a good bit of material ejected from the crucible, but I flooded the room and adjacent hallway with some awful-smelling mixture of Cl
2 and HCl gas. I think that the reaction was essentially complete after just a few hours (if that), as the chlorine stench faded fairly rapidly, though I let the Co
3V
2O
8 run proceed for three days while slowly decreasing the temperature. For the Ni
3V
2O
8 run, I simply loaded the crucible with the reactants, allowed them to dehydrate for a couple hours, ramped the temperature to 950ºC, then came back the next day and turned the furnace off. (I performed this second run on a weekend when the building was more or less deserted.) In retrospect, perhaps I should have used NiO and Co
3O
4 as reactants in the respective runs rather than their chlorides.
Although each reaction proceeded pretty ferociously, I was pleasantly surprised by what I found in the crucible after each run. In each case, the run produced an easily crumbled agglomeration of fine crystals. Although the crystal dimensions are relatively small (up to about 200 microns), the grains are generally still of adequate size for microprobe work. The hardness is moderate, and polishing is easy. In the Co vanadate run, I obtained mostly dark gray, irregular to equant crystals of Co
3V
2O
8 with small amounts of Co
3O
4 and Co
2V
2O
7; inclusions of CoO are present in the Co
3O
4. In the Ni vanadate run, I obtained mostly golden brown, euhedral, prismatic crystals of Ni
3V
2O
8. Some grains have coatings of NiO.
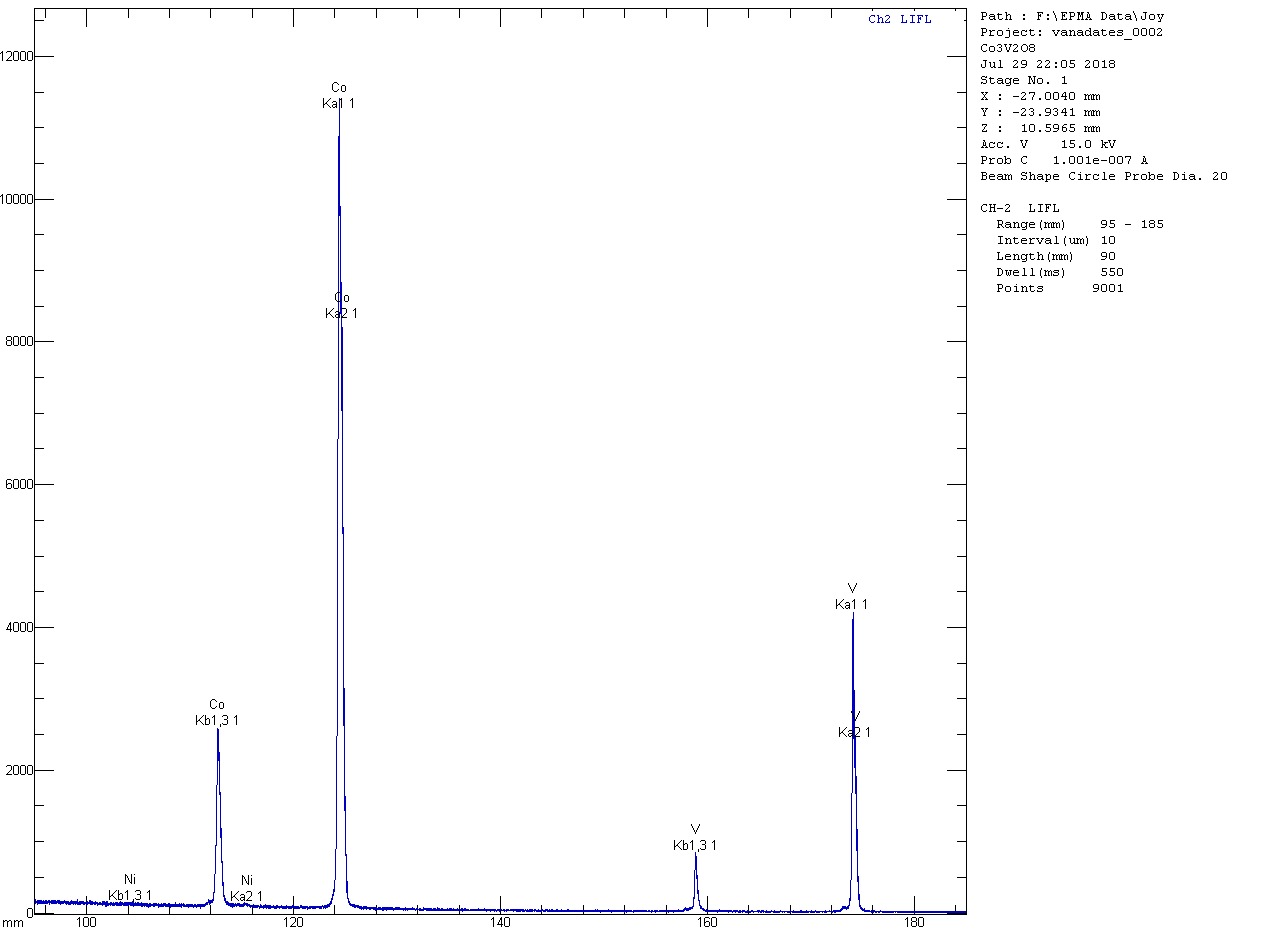
Co
3V
2O
8:

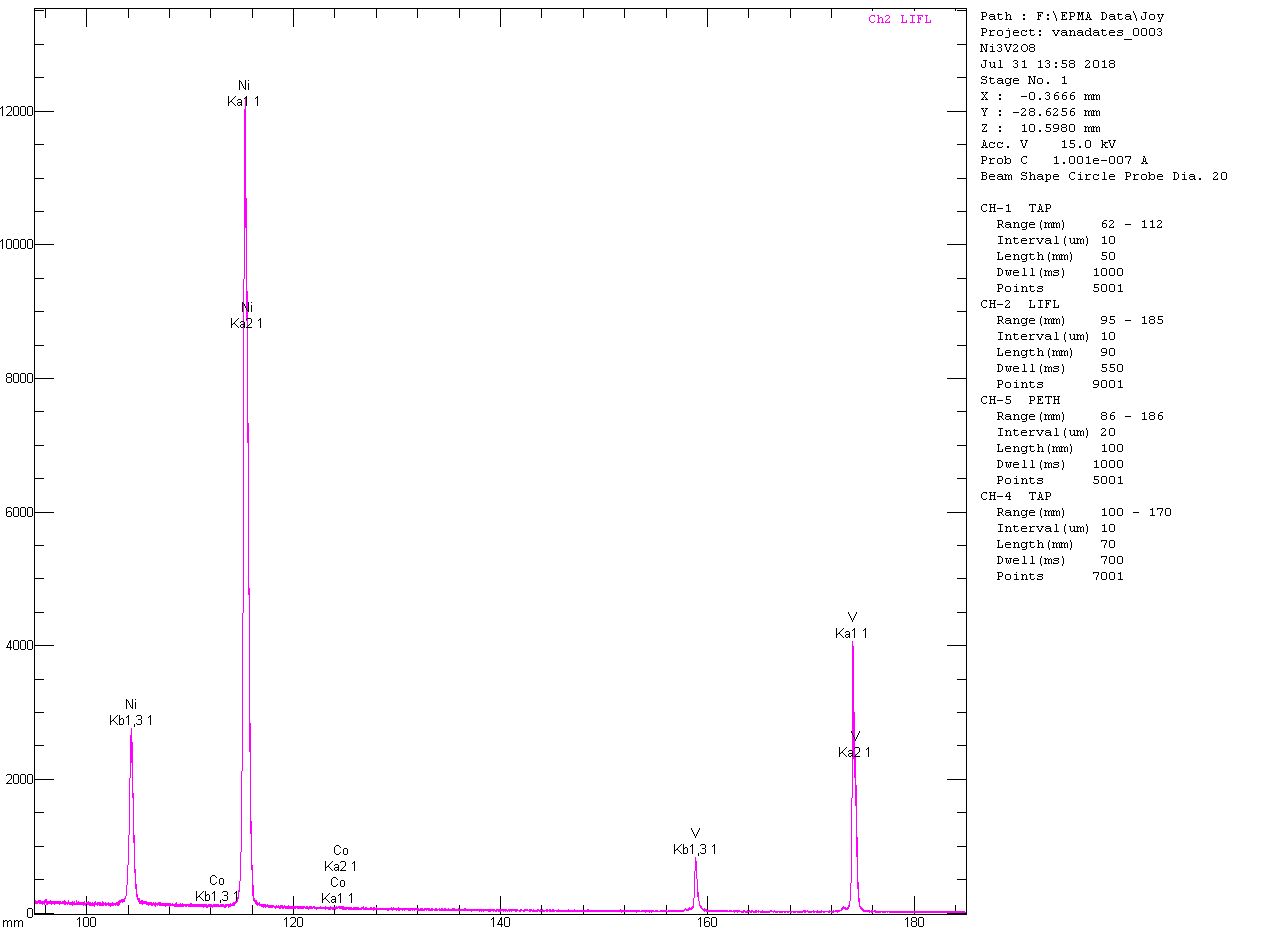
Ni
3V
2O
8:

In wavelength scans, the only impurity I can find in the Co
3V
2O
8 is a small amount of NiO, which averages 0.16 wt% in 30 quantitative analyses and is a bit disappointingly variable, with values ranging from 0.00 to 0.35 wt%. Conversely, I was able to find a small and more uniform amount of CoO in the Ni
3V
2O
8. In analyzing the Ni
3V
2O
8 (32 analyses), I used the Co
3V
2O
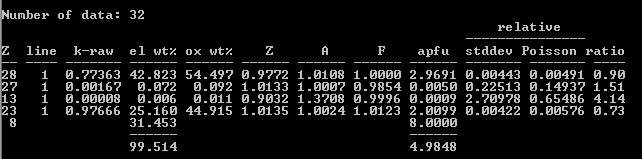
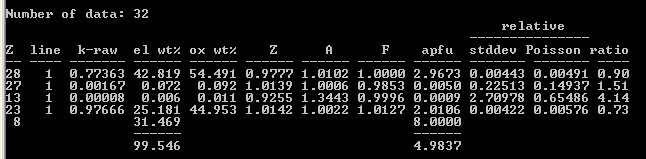
8 as a V standard (and synthetic liebenbergite as Ni standard) and obtained the following results, which I find pretty acceptable at least for V
2O
5. The NiO is low perhaps due to inaccuracy in the atomic number correction; I’ll post analytical results again once I’ve grown some NiO to use as a standard:
PAP/MAC30:
Armstrong/FFAST:
In the attached papers, the authors present flux methods for greatly increasing the sizes of individual crystals of Co
3V
2O
8 and Ni
3V
2O
8 following solid state synthesis. The method requires, following melting, precise temperature ramp control (-0.5ºC/hour) over periods of 400-600 hours not including “several” constant-temperature soak segments incorporated to eliminate melt inclusions.
Although I haven’t done this yet, it is also possible to synthesize Mn
2V
2O
7 (see attached paper, in which the authors appear to have been worried about oxidation of Mn). Analysis of such a compound using pure oxides should be a good test of the fluorescence correction for V Kα, as both Mn Kα and Mn Kβ are able to ionize the K shell of V, but the absorption and atomic number corrections for V Kα should be small.


 To see the latest forum posts scroll to the bottom of the main page and click on the View the most recent posts on the forum link
To see the latest forum posts scroll to the bottom of the main page and click on the View the most recent posts on the forum link