This is a little off topic because I'm not sure how it applies to vacancies in a crystal structure, but it might be worth noting that one can obtain high or low totals in porous materials. But for me the reasons are not entirely obvious and it gets complicated as soon as one starts to think about it. For example, we do know that mere changes in material density do not affect our analytical results *in homegeneous bulk materials* as discussed here:
https://probesoftware.com/smf/index.php?topic=126.msg508#msg508As one real world example, in a low density Al2O3 material I analyzed many years ago, even though it was carbon coated and quite conductive on the polished surface, we obtained totals of around 75% analyzing for Al ka (and assuming Al2O3 stoichiometry) compared to a crystalline Al2O3 material.
It would be difficult to model this using Monte Carlo I suspect, but maybe a simply geometry could be created for Penepma to test for the effect of voids in porous materials?
My hypothesis at the time was that because of the structure of this porous material, there were internal surfaces present (voids) that allowed static charge from the incident electrons to accumulate on the internal surfaces of these pores, thus creating a subsurface charge that caused the incident electron range to be anomalously shallow. This subsurface charging of the voids, I suspect could produce fewer x-ray emissions by increasing the apparent the stopping power of the material. This is assuming that the voids are evacuated and do not have ambient H2O adhering to their surfaces- another "real world" complication.
On the other hand, in a conductive porous material, if the internal surfaces are conductive, the incident electrons should not produce excess sub surface charging, but if they are filled with water or another gas, the fact that the interaction volume is not homogeneous can change the matrix effects significantly. We've all seen how our analytical totals can become anomalously high as the beam traverses a material boundary, even when secondary fluorescence is not an issue. For example a Cu-Al boundary:
https://probesoftware.com/smf/index.php?topic=1064.msg7040#msg7040Think of it this way: at the boundary between pure Cu and pure Al, the instrument is seeing the x-rays from both copper and aluminum, but almost all the Cu Ka x-rays are being emitted from pure copper, and almost all the Al Ka x-rays are being emitted from pure aluminum. So instead of applying a matrix correction of around 1 to most of the emitted x-rays, because our bulk matrix model assumes the sample is roughly a 50:50 composition, we get an severe over-correction for Al Ka because the physics model says that the matrix correction for Cu and Al in an alloy is around 1 for Cu Ka, but around 1.5 for Al Ka! This may be part of the reason why we get this odd looking *totals* map in a Cu-Al eutectic map:
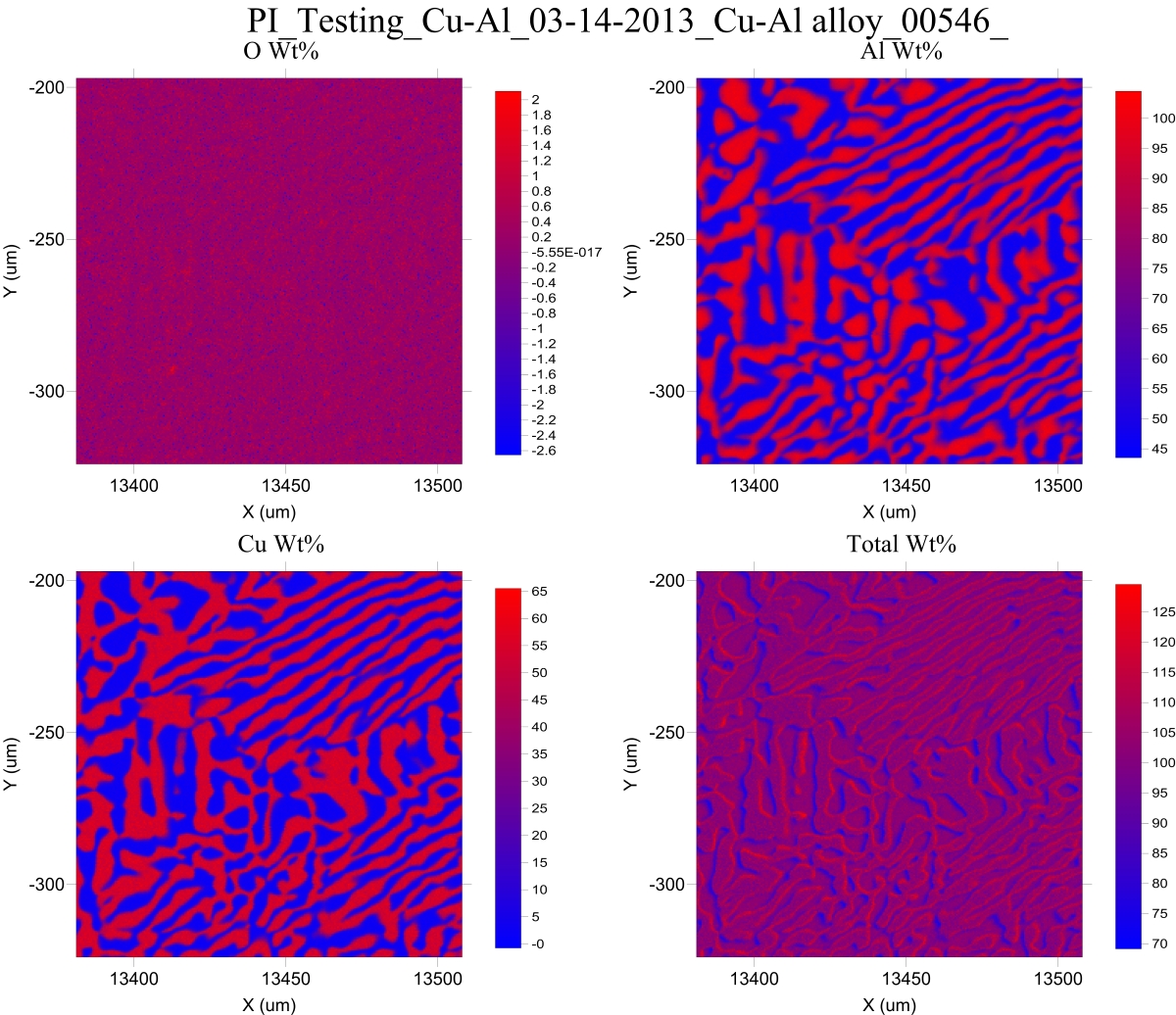
Now of course these effects are very sensitive to detector orientation but I suspect that at least part of what we are seeing above is due to an inhomogeneous interaction volume.
Of course in the case of a highly self fluorescing system, one can even get *low* totals when the self fluorescing material is bounded by a material that does not contain the element causing the fluorescence in a self fluorescing material:
https://probesoftware.com/smf/index.php?topic=58.msg223#msg223It gets complicated real fast when the interaction volume is not homogeneous. As Chuck Fiori once said: "If the interaction volume is not homogeneous, all bets are off"!
The problem is on what scale does the inhomogeneity become important? Clearly it doesn't matter at the atomic scale or we couldn't measure compounds. So somewhere between the scale of atoms and the interaction volume it begins to matter, and of course that scale depends on the beam energy and the other physics details.
Someone should look into this stuff...


 You cannot see "members only" boards if you are not a member, so please join the forum!
You cannot see "members only" boards if you are not a member, so please join the forum!