I was recently chatting with a colleague and we were lamenting the situation where we sometimes encounter a user who just hasn't kept up with the EPMA literature. In this case it's the quantitative spectral interference correction in Probe for EPMA which has been implemented since the 1990s! For details see this paper published in 1993 (the pdf is attached below):
J. J. Donovan, D. A. Snyder and M. L. Rivers, "An Improved Interference Correction for Trace Element Analysis" Microbeam Analysis, 2: 23-28, 1993
Of course it doesn't help that these older issues of Microbeam Analysis do not show up in searches of Google Scholar! Therefore it's important to keep in mind that some science did occur before the Internet was invented!

But for those who learned about EPMA in the 1990's from say, people who may have learned EPMA in the 1970s or 1980s, this can be of concern... because apparently some people are unaware that one can indeed quantitatively correct for spectral interferences in EPMA.
So I thought I would start with the example that had been raised by a user who claimed "you can't analyze for minor/trace vanadium in titanium bearing minerals", e.g., ilmenites.
Now we're just looking at standards here today, since we can be sure we know what their actual vanadium contents are (though these interference corrections work equally well on unknown samples), but since I didn't have an ilmenite standard handy, I used an SrTiO3 standard as a proxy. So after tuning up with Ti metal, V metal and SrTiO3 for Ti Ka, V Ka and Sr La respectively, we next perform wavescans on each standard just to observe the spectra in this region:
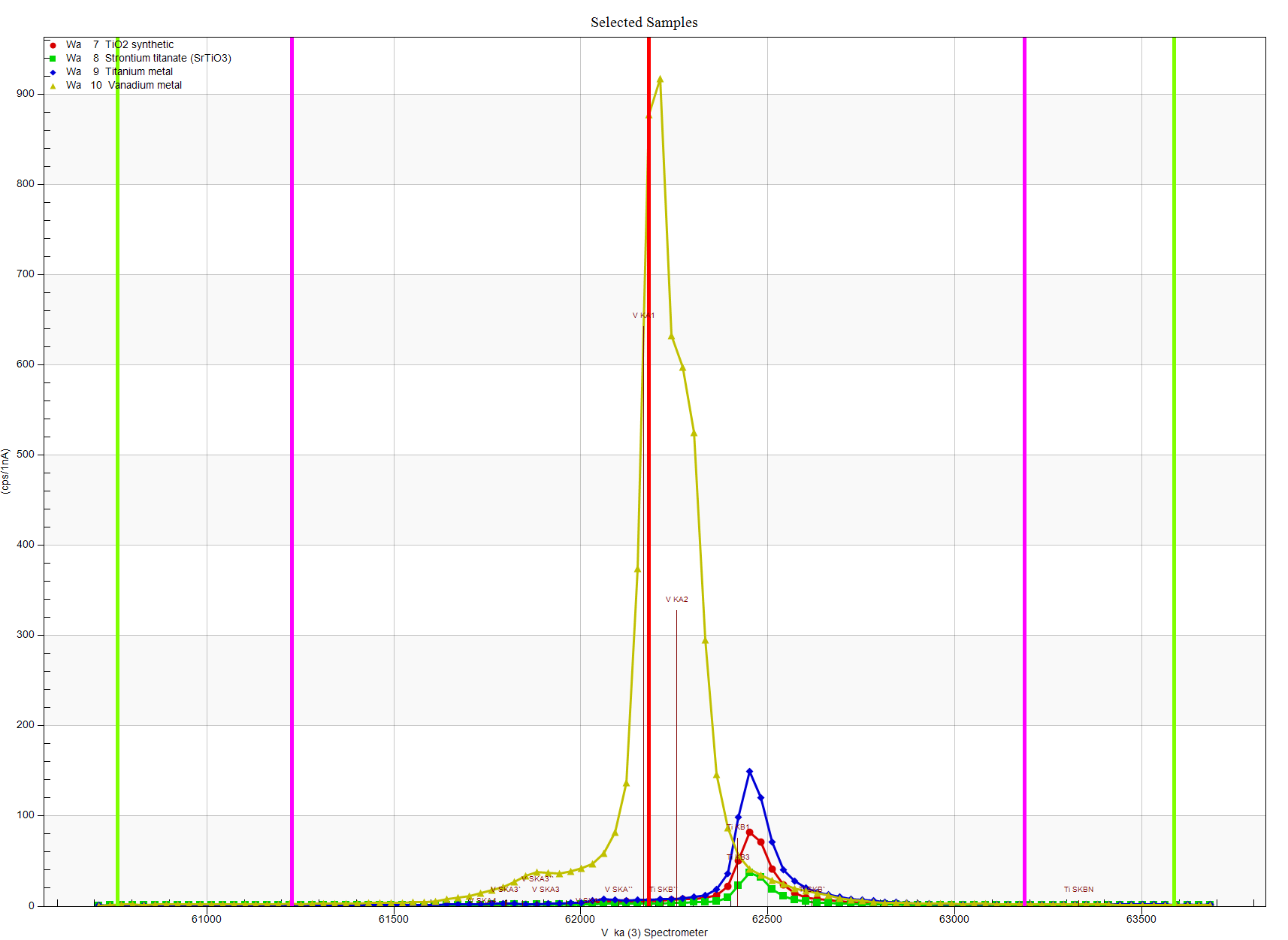
Clearly the extended tail of the Ti Kb line is interfering with the V Ka line as expected. Zooming in to see the background situation more closely we see this:
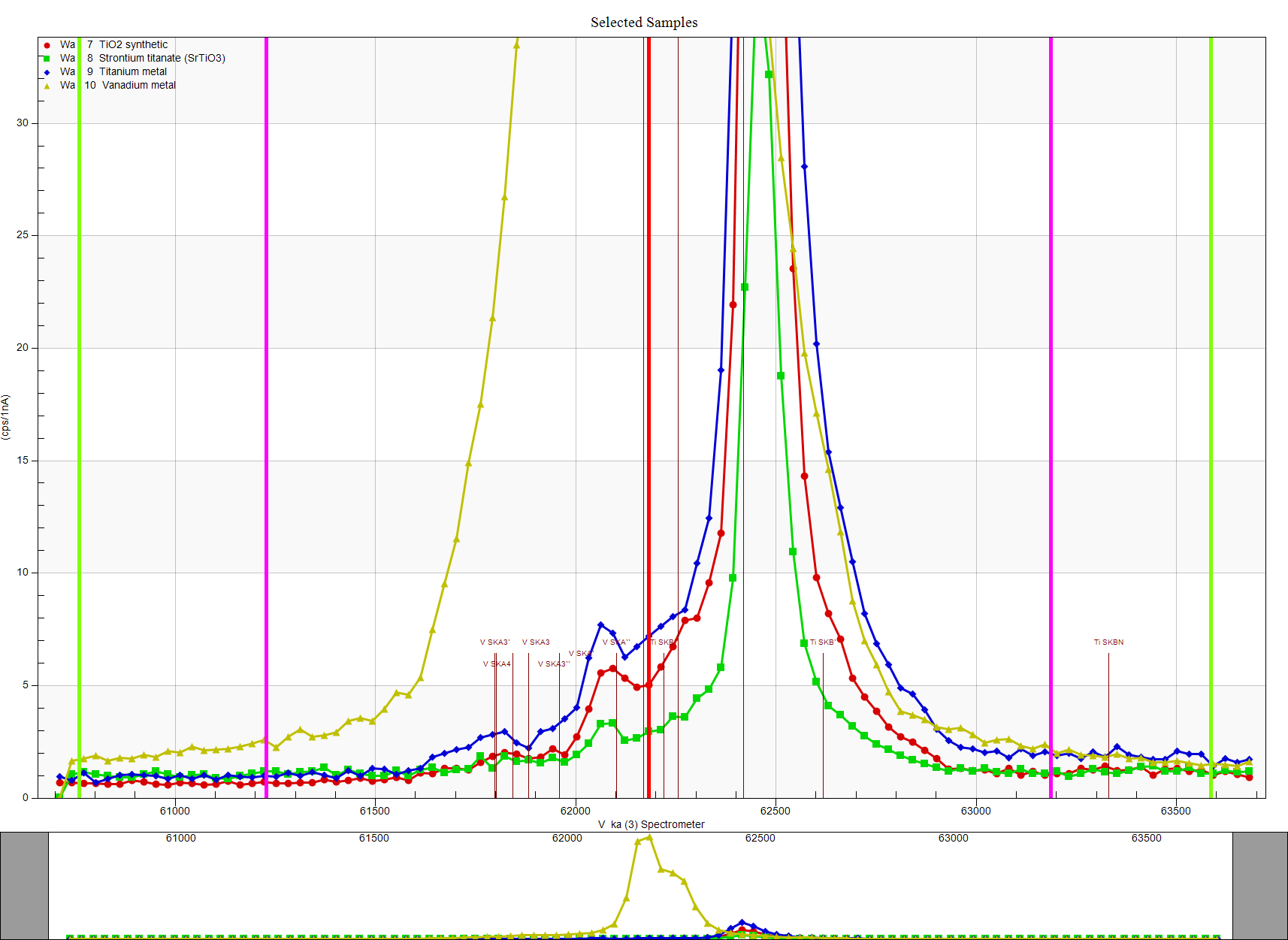
Note that the default positions for the high and low off-peak positions (magenta vertical lines) have been adjusted to be further away (green vertical lines) from the "tails" of the emission peaks. These tails of course are due to polygonization treatment of the Bragg crystals after bending and plastic deformation in order to improve reflectivity. See here for more details on this:
https://probesoftware.com/smf/index.php?topic=79.msg7818#msg7818This adjustment of the backgrounds is quite important for trace elements when a large interference is present, because we are essentially going to be subtracting two large numbers from each other to obtain a small number. That reminds me to also mention another option in PFE to be sure that one is measuring the interference calibration with the same sensitivity as used for the unknown sample, as shown here:
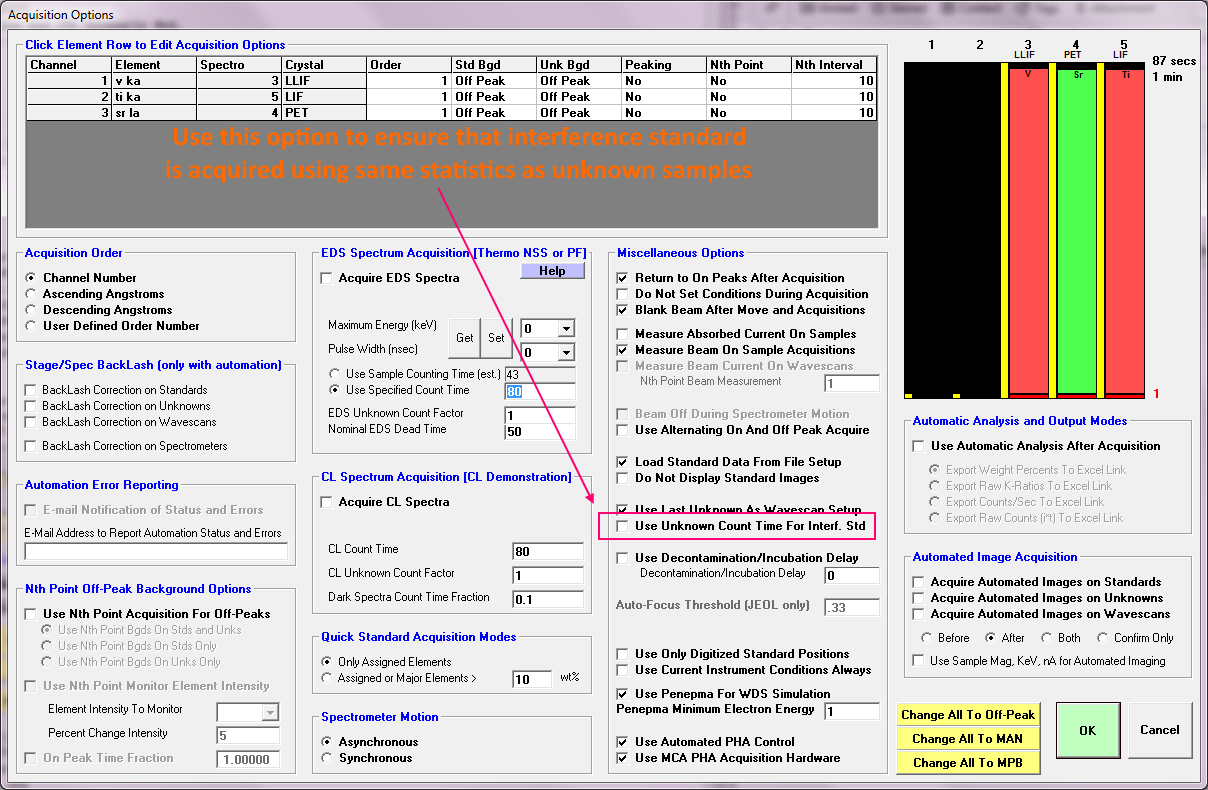
And remember, for the quantitative interference correction, we must measure *both* the interfered emission line, and also the interfering emission line. Furthermore, we must use a standard for the interference correction calibration that contains a known amount of the interfering element (Ti) and *none* of the interfered element (V). So we must measure at least V Ka and Ti Ka and can use TiO2 as a standard for the interference calibration, assuming it has no V as a contaminant.
So, let's first see what the magnitude of this interference is quantitatively without an interference correction when measuring V in TiO2:
ELEM: V Ti Sr O SUM
855 .476 59.984 -.002 40.050 100.507
856 .468 59.860 -.006 40.050 100.372
857 .475 59.737 -.015 40.050 100.248
858 .489 60.108 .008 40.050 100.654
859 .484 59.878 -.029 40.050 100.383
AVER: .478 59.913 -.009 40.050 100.433
SDEV: .008 .139 .014 .000 .154
SERR: .004 .062 .006 .000
%RSD: 1.74 .23 -155.15 .00
PUBL: n.a. 59.939 n.a. 40.050 99.989
%VAR: --- (-.04) --- .00
DIFF: --- (-.03) --- .000
STDS: 523 22 251 ---
So almost 5000 PPM, which is pretty bad, right? Now we apply the interference correction in PFE from the Standard Assignments dialog as seen here:
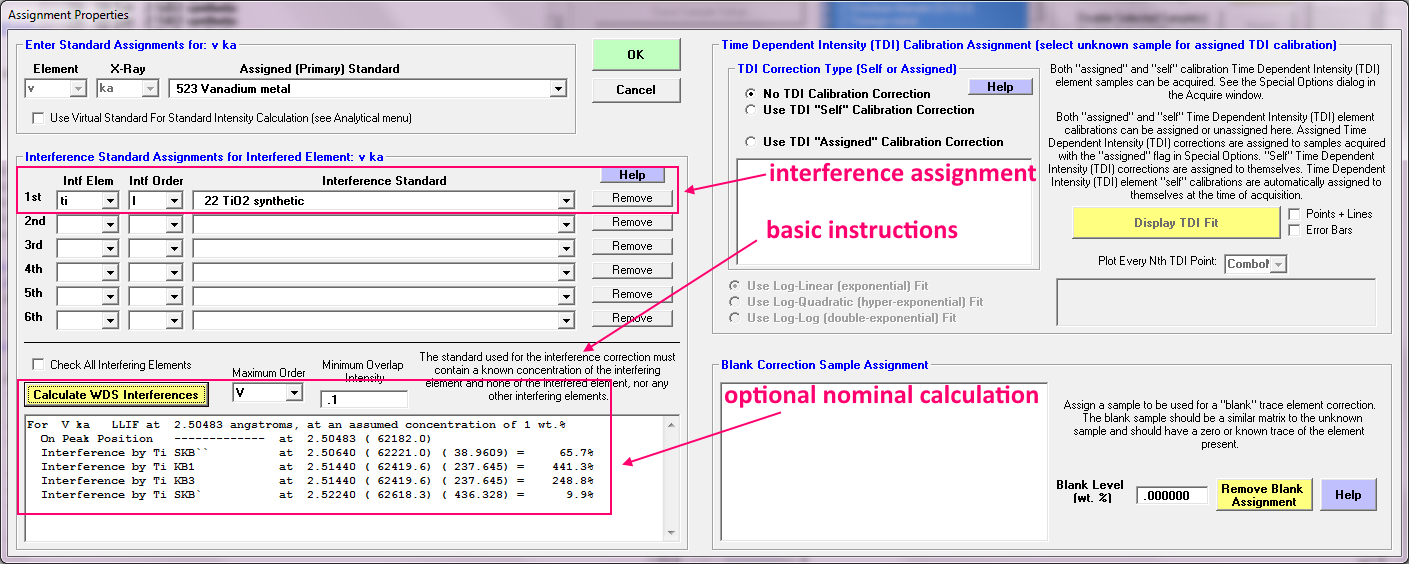
The actual interference assignment is in the upper red rectangle, the optional nominal calculation assuming a Gaussian peak shape is in the lower red rectangle and used only to decide if the apparent interference is due to an actual interference or merely contamination in the standard.
With the interference correction applied we obtain this value for V Ka in TiO2:
ELEM: V Ti Sr O SUM
855 -.003 60.008 -.002 40.050 100.052
856 -.010 59.884 -.006 40.050 99.918
857 -.001 59.761 -.015 40.050 99.795
858 .009 60.132 .008 40.050 100.199
859 .006 59.902 -.029 40.050 99.929
AVER: .000 59.937 -.009 40.050 99.978
SDEV: .008 .139 .014 .000 .153
SERR: .003 .062 .006 .000
%RSD: ---- .23 -155.15 .00
PUBL: n.a. 59.939 n.a. 40.050 99.989
%VAR: --- (.00) --- .00
DIFF: --- (.00) --- .000
STDS: 523 22 251 ---
Now our apparent V concentration is of course zero, simply because this is the standard utilized for the interference calibration. Now let's apply this same interference calibration to a sample with a very different matrix, SrTiO3, first without an interference correction:
ELEM: V Ti Sr O SUM
830 .199 26.105 47.632 26.154 100.090
831 .205 26.169 47.918 26.154 100.446
832 .207 26.253 47.718 26.154 100.332
833 .207 26.262 47.751 26.154 100.375
834 .206 26.371 47.789 26.154 100.520
AVER: .205 26.232 47.762 26.154 100.352
SDEV: .003 .101 .105 .000 .163
SERR: .001 .045 .047 .000
%RSD: 1.54 .38 .22 .00
So an apparent vanadium concentration of ~2000 PPM! And now *with* the interference correction:
ELEM: V Ti Sr O SUM
830 -.010 26.111 47.618 26.154 99.873
831 -.005 26.175 47.904 26.154 100.228
832 -.004 26.259 47.704 26.154 100.113
833 -.003 26.268 47.737 26.154 100.156
834 -.006 26.377 47.775 26.154 100.300
AVER: -.005 26.238 47.747 26.154 100.134
SDEV: .003 .101 .105 .000 .162
SERR: .001 .045 .047 .000
%RSD: -50.13 .38 .22 .00
PUBL: n.a. 26.103 47.742 26.154 99.999
%VAR: --- .52 (.01) .00
DIFF: --- .135 (.01) .000
STDS: 523 22 251 ---
Now our apparent vanadium content is around -50 PPM which is a slight over correction, but still within 2 sigma (60 PPM) of zero, so we cannot really say that this is statistically significant with 99% confidence. In fact the 99% confidence detection limit for this vanadium in this matrix is around 140 PPM!
Actually I'm pretty sure that this -50 PPM offset from zero is due to the fact that I (unthinkingly) tuned up Ti Ka on Ti metal, and we are looking at Ti oxides, and there is a tiny peak shift from the metal to the oxide. I'll try running it again again, next time tuning up Ti Ka on TiO2 and let you all know how that goes.
But really when you think about it, -50 PPM is certainly more accurate (closer to zero) than roughly 2000 PPM! And considering that that is about a 4000% correction, it's really not too bad!



 Please keep your software updated for best results!
Please keep your software updated for best results!