A very nice tutorial for quantitative calculations in CalcZAF can be performed using the default CalcZAF.dat input file. The following post is a step by step exercise discussing the various data types in this input file and options for the quantitative calculations.
If you do not have CalcZAF already installed on your computer, you can download it for free here:
https://www.probesoftware.com/download/CalcZAF.msiAssuming you already have CalcZAF installed, you should first check that it is up to date by running the Help | Update CalcZAF menu and clicking the Download Update button and following the on-screen directions.
When finished updating CalcZAF we can start by running CalcZAF.exe, and clicking the File | Open menu and selecting the default CalcZAF.DAT file as seen here:

Note that the format of this file is documented in the Probe for EPMA User Reference manual which can be accessed by hitting the F1 key in CalcZAF. You can also output CalcZAF format files from Probe for EPMA for test purposes. The first "sample" which is MgO will be loaded to the Calculate ZAF Corrections window as seen here:

Note that the first calculation option "Calculate Intensities From Concentrations" is automatically selected because that is specified in the CalcZAF.dat file. Note that if you enter a composition from a formula or the standard database, this is the default option for calculation. Next click the Calculate button and the results will be output to the ZAF Calculations and log windows as seen here:
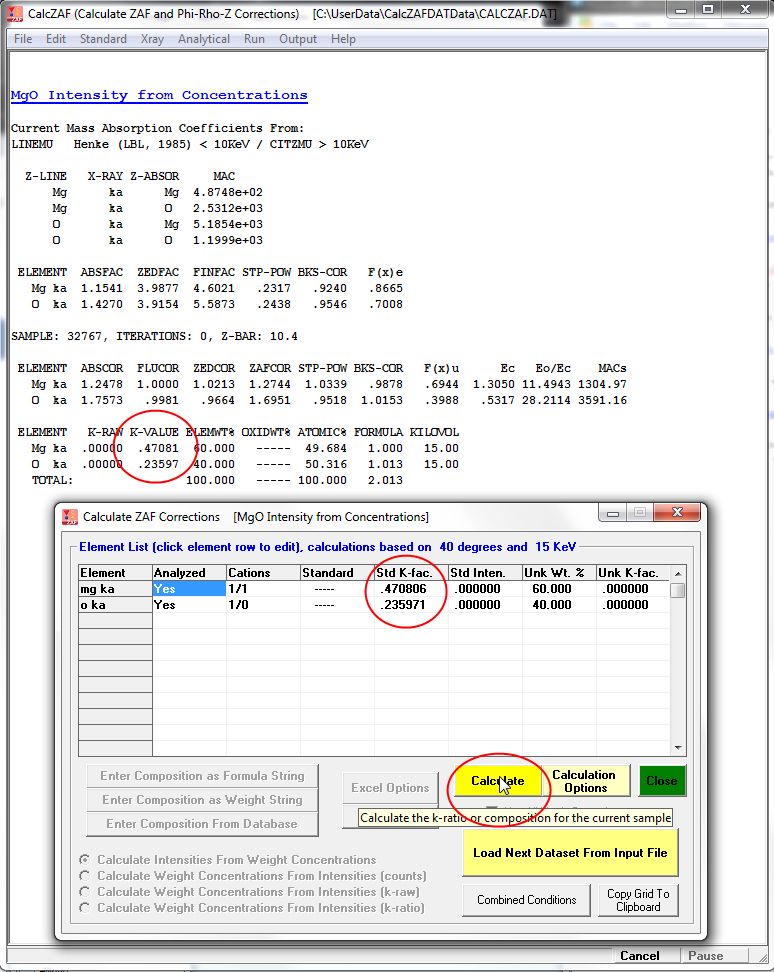
Note the calculated k-ratios along with a number of other useful parameters. You can click on the element row to change the element concentrations or the Analytical | Operating Conditions menu to change the keV and then click the Calculate button again as desired.
We can now load the next "sample" in the CalcZAF.Dat file by clicking the Load Next Dataset From Input File as shown here:

and see that it is a calculation of concentrations from Fe, Si and O unknown *and* standard intensities and of course the standard compositions. When the Calculate button is again clicked we see the following results:
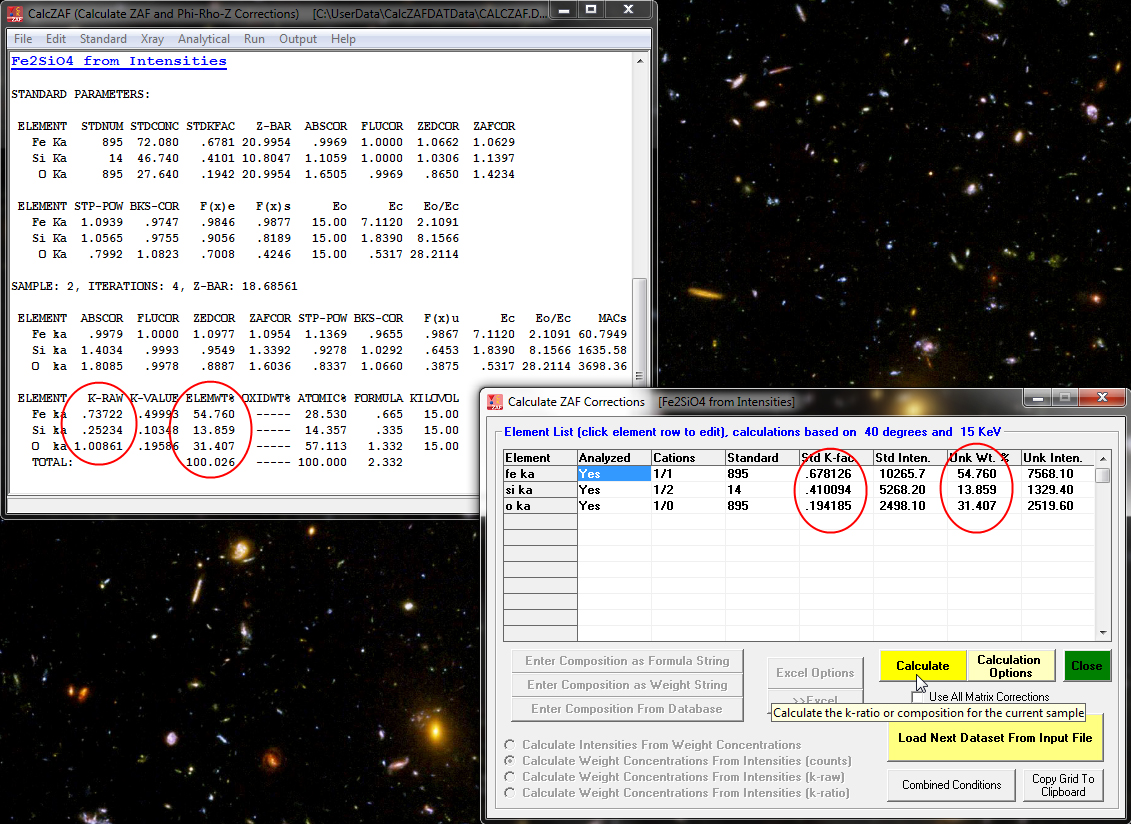
Now an interesting option is the ability to compare the results of the 10 different matrix corrections (and 6 different mass absorption coefficient tables) in CalcZAF- courtesy of John Armstrong who wrote the original CITZAF code. This is performed by simply checking the Use All Matrix Corrections checkbox shown here and again clicking the Calculate button:
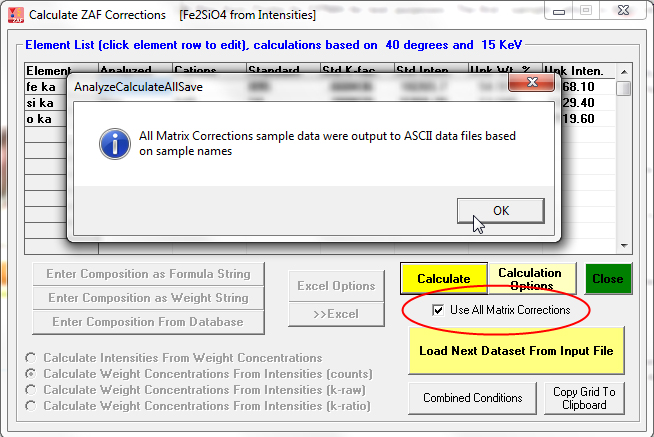
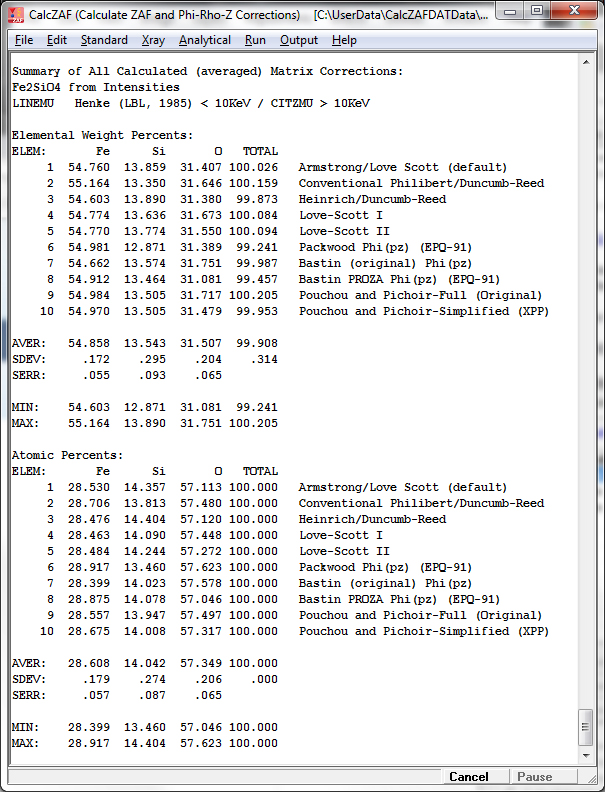
The tabulated results can be seen listed in the log window after clicking OK and then exporting the results to Excel (if available):
The next sample is an alloy steel, again unknown and standard intensities with pure element standards:

This next sample is an iron oxide calculated from raw k-ratios and standard compositions only:
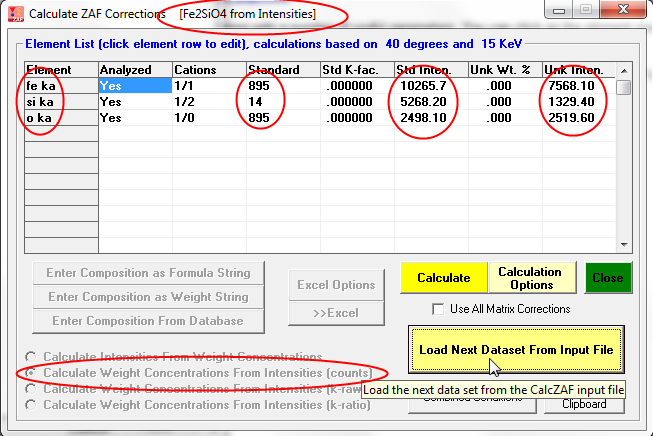
This example is again MgO, but compositions calculated from unknown k-ratios normalized to pure elements:
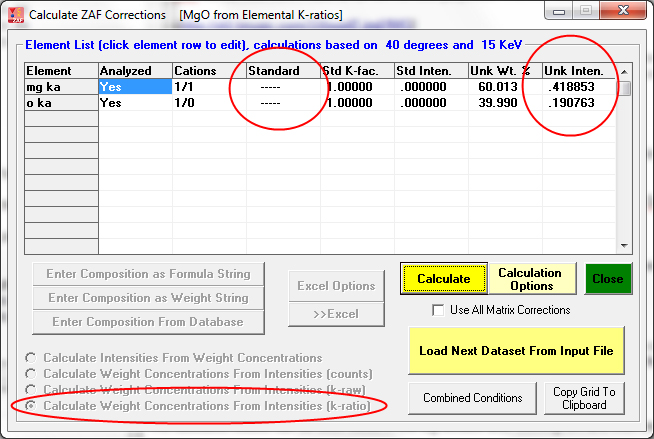
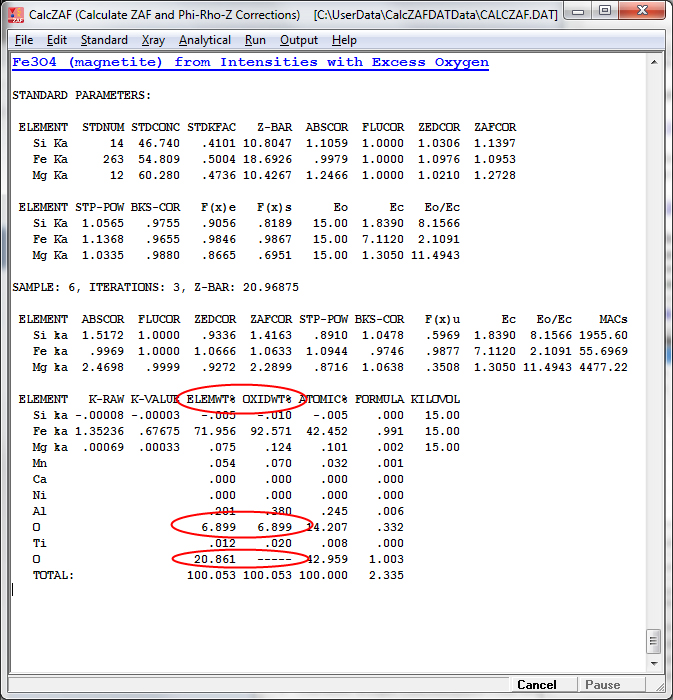
This next sample is magnetite (Fe3O4) calculated from unknown and standard counts (and standard compositions) with oxygen calculated by stoichiometry:

The excess oxygen when Fe is displayed as FeO is visible here:
This next example is Si calculated by difference. The calculation by difference and other calculation options in CalcZAF are accessed with the Calculation Options button shown here:
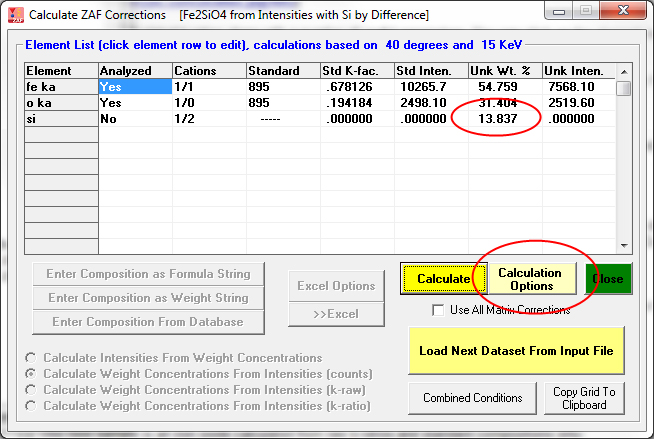
This is a slightly more complicated calculation where oxygen is calculated by stoichiometry and Fe is calculated by stoichiometry to the stoichiometric oxygen. Therefore the only element actually measured in this example was Si!

Note that the above type of calculation is often applied to carbonates where only the cations (Ca, Mg, Fe, Mn, etc) are measured, oxygen is calculated by stoichiometry and carbon is calculated by stoichiometry to the calculated oxygen by the rule 0.333 carbon atoms for each oxygen atom.
The remaining examples are similar to what we have already looked at above, but a few others deserve additional explanation. This example of calculating stoichiometric oxygen in a matrix where some of the oxygen is replaced by a halogen element is interesting:

Because the matrix correction was calculated with the assumption of stoichiometric oxygen as shown above, the absorption correction of F ka by oxygen is over calculated. Particularly since the absorption of F ka by oxygen is quite severe. However, by simply applying a correction for oxygen equivalent of halogen in the matrix iteration we can rigorously calculate the proper absorption correction for F ka by stoichiometric oxygen as seen here:

The modified absorption correction and results are seen here and it is worth noting that without the subtraction of the oxygen equivalent of fluorine from the calculated oxygen during the matrix correction, the fluorine values will be in error by over 20% relative (9.252 wt% without the halogen equivalent correction and 9.023 wt% with the halogen equivalent correction in the matrix correction)!
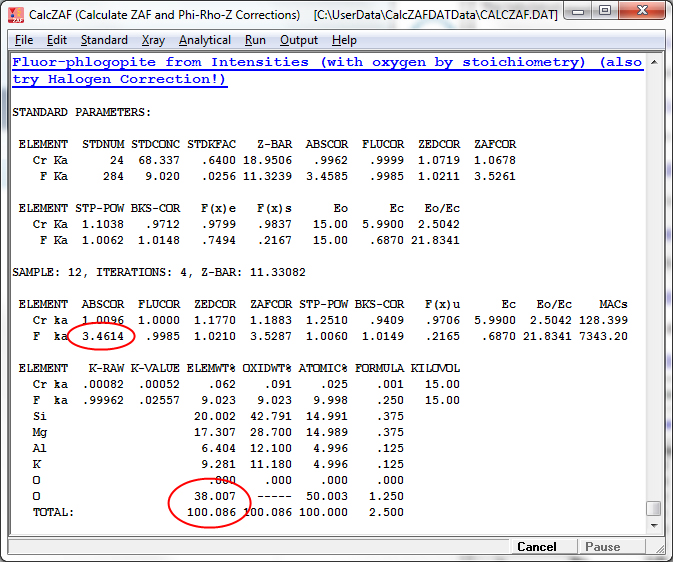
Finally note that the standard used for fluorine should be entered properly into the standard composition database (Standard.mdb) with the oxygen equivalent of halogen applied as seen here:

There are many other calculation options available in CalcZAF, but this post should get you started. As always, please let us know if you have any questions related to this post.
A couple of documents that you might find useful:
http://epmalab.uoregon.edu/pdfs/JTA-1988-ZAF.pdfhttp://epmalab.uoregon.edu/pdfs/CalcZAF,%20TryZAF%20and%20CITZAF.pdf

 To post in-line images, login and click on the Gallery link at the top
To post in-line images, login and click on the Gallery link at the top