As previously mentioned,
http://probesoftware.com/smf/index.php?topic=114.msg3322#msg3322I decided to acquire some data to see if we could quant carbon using the MAN correction on metals. First I had Julie polish and Ag coat one of our standard blocks with about 15 nm of Ag. I then analyzed C, N, Mo, Fe and Si in pure C, AlN, Mo, Fe and Si (for a range of x-ray energies and average Zs), at 10 keV and 50 nA (for improved sensitivity). I counted 80 seconds on each data point (on-peak MAN only for carbon and nitrogen) and used 16 TDI intervals ( 5 sec each).
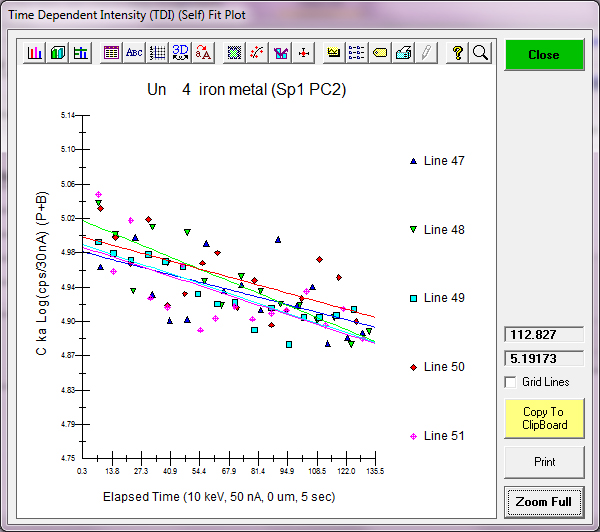
First of all, just as shown in the previously acquired data above, the carbon signal shows a small *decrease* over time while all other elements remain stable within precision:
Here is nitrogen Ka:

As for the quant, here you can see that results on a pure Fe std (but acquired separately from the MAN background stds), gives zero within precision for all elements (except for Si at 230 PPM which could be due to the colloidal Si polishing) :
Un 4 iron metal, Results in Elemental Weight Percents
ELEM: C N Mo Fe Si
BGDS: MAN MAN LIN LIN EXP
TIME: 80.00 80.00 60.00 60.00 60.00
BEAM: 49.75 49.75 49.75 49.75 49.75
ELEM: C N Mo Fe Si SUM
47 -.023 -.018 -.025 100.090 .023 100.047
48 .032 .037 .005 100.471 .025 100.570
49 -.010 -.043 -.006 101.030 .021 100.992
50 .005 .006 -.004 100.380 .026 100.414
51 -.013 -.030 -.031 100.456 .021 100.404
AVER: -.002 -.010 -.012 100.486 .023 100.485
SDEV: .021 .032 .015 .341 .002 .342
SERR: .010 .014 .007 .152 .001
%RSD: -1244.94 -331.31 -125.84 .34 10.30
STDS: 506 672 542 526 514
STKF: .9705 .1485 .9901 1.0000 1.0000
STCT: 23361.3 459.1 4349.2 1085.6 73411.0
UNKF: .0000 -.0001 -.0001 1.0048 .0002
UNCT: -.2 -.2 -.5 1090.8 15.0
UNBG: 148.0 13.6 9.2 7.9 134.1
ZCOR: 2.4870 1.5301 1.1661 1.0000 1.1457
KRAW: .0000 -.0004 -.0001 1.0048 .0002
PKBG: 1.00 .99 .95 139.30 1.11
INT%: ---- ---- ---- ---- ----
TDI%: 5.364 -.141 ---- ---- ----
DEV%: .6 3.2 ---- ---- ----
TDIF: LOG-LIN LOG-LIN ---- ---- ----
TDIT: 131.80 134.00 ---- ---- ----
TDII: 148. 13.3 ---- ---- ----
TDIL: 5.00 2.59 ---- ---- ----
I also performed some "fast" beam scans in Probe Image *on the same spot* on pure Fe and will examine that data next week for TDI effects, but in the meantime here is my question: is the small decrease in carbon signal over time, something that is observed on other instruments?
I would have thought that the carbon signal would increase over time, not decrease... so I have to wonder if the cryo baffle (~100 Kelvin) on my instrument is somehow related to this? I would be very grateful for similar measurement results by other labs... thanks!
Again this is a freshly polished Fe metal sample coated with 15 nm Ag and measured at 10 keV and 50 nA. In any case, a standard deviation of around 200 PPM for carbon in 80 seconds isn't bad. That is, with a 5% TDI correction (+/- 0.6%). Which would correspond to an absolute TDI correction of ~800 +/- 100 PPM concentration.
Finally, is this decrease in carbon intensity simply related to the so called "decontamination time", as coined by Stuart Kearns, et al.
http://probesoftware.com/smf/index.php?topic=48.msg161#msg161Though I would have expected the decontamination effect to apply only to the first 10 seconds or so, at most?


 To post in-line images, login and click on the Gallery link at the top
To post in-line images, login and click on the Gallery link at the top